Как матери детей с синдромом прадера
Содержание:
- Ваня выздоровел
- Лечение ЗПРР при хромосомных заболеваниях.
- Другие симптомы
- Диагностика
- Распространённые патологии при синдроме Дауна
- Генетика
- Благотворительные фонды помогают приехать на лечение в Израиль
- Диагностика ожирения
- Если говорить простым языком, что такое синдром Прадера — Вилли?
- Интересные факты о синдроме
- Общие сведения о синдроме Вилли-Прадера
- Диагностика СПВ
- Причины
- Генетика
- Синдром Прадера-Вилли
Ваня выздоровел
В Украине Ване Волохатюку в течение 2 лет не могли поставить диагноз. В МЦ Хадасса у него обнаружили редкое и очень опасное заболевание – синдром IPEX. Ребенка спасли, проведя успешную трансплантацию костного мозга (ТКМ).
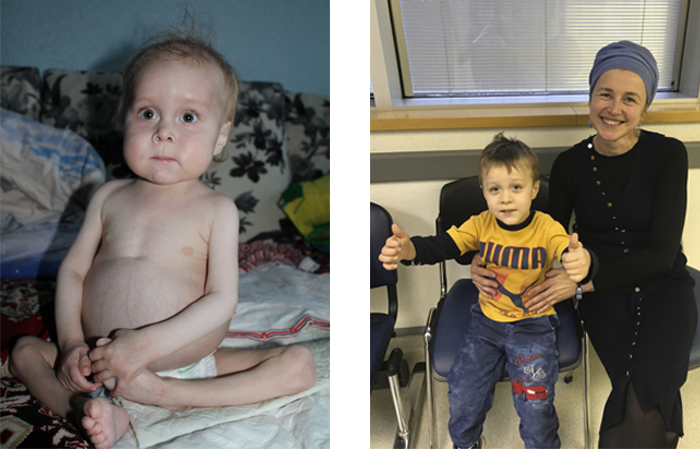
Фото слева: так Ваня выглядел до того, как профессор Полина Степенски успешно провела ему ТКМ в медицинском центре Хадасса.
Фото справа: Ваня после ТКМ с сотрудницей нашего медицинского департамента Эстер Винокуров.
Для справки:
- По разным оценкам в России к 2017 году было зарегистрировано от 1.5 до 5 миллионов больных с редкими заболеваниями.
- В 2017 году в Украине на лечение орфанных заболеваний был выделен 1 млрд. гривен, и эта сумма почти сразу же была признана недостаточной.
- В Беларуси зарегистрированы тысячи случаев орфанных заболеваний, по оценкам местной прессы ими болеют от 6% до 8% населения.
Лечение ЗПРР при хромосомных заболеваниях.
Основой лечения является уникальная методика патогенетической терапии речевых расстройств при хромосомной патологии — биофизическая активация нейромоторных структур, основу которого составляет щадящая стимуляция проводников нервной системы микротоками с использованием нейрофизиологического прибора. Метод лечения базируется как на активации самих речевых центров, так и на восстановлении нарушенных связей между центрами и полушариями головного мозга. Помимо этого, восстанавливаются разрозненные связи речевых центров с другими областями мозга, участвующими в реализации речевой функции. В процессе лечения формируется физиологичное, последовательное взаимодействие всех зон мозга, связанных с речепродукцией. В результате появляется речь.
Проведение биофизической активации сочетается с дополнительными методиками лечения, такими как — лимфомежклеточная терапия, которая применяется для регулирования интегративной деятельности и восполнения дефицита энергетической системы мозга и позволяющая применять малые дозы церебропротекторов, которые вводятся эндолимфатически и попадают в ткани головного мозга, минуя гематоэнцефалический барьер.
В качестве другого способа использования препаратов с нейротрофическим и антиоксидантным действием применяется методика эндоназального электрофореза кортексина, что позволяет вводить лекарственные препараты непосредственно в ткани головного мозга.
Исследования последних десятилетий выявили, что у большинства детей с речевыми и поведенческими проблемами в различной степени нарушены функции мозжечка и базальных ганглиев. Именно функционирование мозжечка определяет успешность ребенка в обучении. С этой целью применяется уникальная разработка Центра авиакосмической медицины — подошвенный имитатор опорной нагрузки «Корвит», применяемый для нейрофизиологической регуляции стато-кинетической функции ЦНС. В основе терапевтического воздействия аппарата «Корвит» лежит процесс активации опорной афферентации, отвечающей за нормализацию процессов возбуждения и торможения в центральной нервной системе, что приводит к уменьшению спастичности мышц, развитию и закреплению функциональных связей в головном мозге, способствующих восстановлению координации движений, и, опосредованно, улучшению речи и мышления.
Также для успешного лечения различных форм ЗПРР специалистами применяется одно из достижений современной науки — метод аудиовокальной терапии RUSTOMATIS. Прибор использует звукозаписи высокочастотных и низкочастотных компонентов. При чередовании такой музыки путем напряжения и расслабления у ребенка тренируется аппарат среднего уха – молоточек и стремечко, с помощью чего расширяется диапазон восприятия внешних факторов, увеличивается концентрация внимания, в мозг поступает новая информация и, как следствие исчезают многие нарушения и расстройства.
Обязательным звеном в лечебном комплексе у детей с наличием речевых расстройств является занятия с клиническим психологом, а также логопедическая коррекция, которая включает диагностику степени нарушений, ежедневные занятия, направленные на улучшение речевой функции и логопедический массаж для коррекции различных видов дизартрии и дисфагии.
На фоне сочетания проведения биофизической активации со вспомогательными методиками лечения наблюдаются положительные изменения, которые могут быть видны уже через несколько процедур, но максимальный эффект развивается через полтора-три месяца после курса. Как правило, для закрепления полученных результатов и дальнейшего развития двигательных и когнитивных навыков специалистами центра рекомендуется повторный курс лечения через 5-6 месяцев.
Другие симптомы
В дальнейшем данная патология характеризуется следующими симптомами:
- Искривление позвоночного столба.
- Кариес молочных зубов и повышение густоты слюны.
- Склонность к перееданиям.
- Гипофункции половых желез, которые в дальнейшем приводят к бесплодию.
- Ожирение.
- Задержка моторики и речевого развития.
- Отсталость в психомоторном развитии.
- Запоздалость в половом созревании.
Данные симптомы определяются визуально. В подростковый период выявляются следующие симптомы:
- Задержка речевых навыков.
- Избыточный вес при очень низком росте.
- Неестественная гибкость тела.
- Снижение интеллекта и неспособность к обучению.
Диагностика
Он традиционно характеризуется гипотонией, низким ростом, гиперфагией, ожирением, поведенческими проблемами (в частности, поведением, подобным обсессивно-компульсивному расстройству ), маленькими руками и ногами, гипогонадизмом и легкой умственной отсталостью. Однако при ранней диагностике и раннем лечении (например, с помощью терапии гормоном роста) прогноз для людей с СПВ начинает меняться. Как и аутизм, СПВ представляет собой расстройство спектра, симптомы которого могут варьироваться от легких до тяжелых и могут меняться на протяжении всей жизни человека. Поражаются различные системы органов.
Традиционно СПВ диагностировали на основании клинических проявлений. В настоящее время синдром диагностируется с помощью генетического тестирования; Рекомендуется тестирование новорожденным с выраженной гипотонией. Ранняя диагностика СПВ позволяет своевременно вмешаться и назначить гормон роста на ранней стадии . Детям с СПВ показаны ежедневные инъекции рекомбинантного гормона роста (ГР). GH поддерживает линейный рост и увеличение мышечной массы, а также может уменьшить озабоченность едой и прибавку в весе.
Основой диагностики является генетическое тестирование , в частности, тестирование метилирования на основе ДНК для выявления отсутствия отцовской области PWS / AS на хромосоме 15q11-q13. Такое тестирование выявляет более 97% случаев
Тестирование, специфичное для метилирования, важно для подтверждения диагноза СПВ у всех людей, но особенно у тех, кто слишком молод, чтобы проявлять признаки, достаточные для постановки диагноза на клинических основаниях, или у тех людей, у которых есть атипичные результаты.
PWS часто ошибочно диагностируется как другие синдромы из-за того, что многие в медицинском сообществе не знакомы с ним. Иногда его ошибочно принимают за синдром Дауна просто из-за относительной частоты синдрома Дауна по сравнению с PWS.
Распространённые патологии при синдроме Дауна
В дополнение к особенностям внешнего вида есть увеличенный риск возникновения ряда медицинских проблем:
Гипотония
Почти у всех пострадавших младенцев слабый мышечный тонус (гипотония), что означает, что их мышцы ослаблены и кажутся несколько гибкими. При данной патологии ребёнку будет сложно научиться перекатываться, сидеть, стоять и говорить. У новорождённых гипотония также может вызывать проблемы с кормлением.
Вследствие гипотонии у многих детей задерживается развитие моторных навыков и возможно возникновение ортопедических проблем. Её нельзя излечить, но физическая терапия может помочь улучшить мышечный тонус.
Нарушения зрения
Проблемы со зрением распространены при синдроме и есть вероятность того, что они будут увеличиваться с возрастом. Примерами таких расстройств зрения являются близорукость, дальнозоркость, косоглазие, нистагм (непроизвольные движения глаз с высокой частотой).
У детей с трисомией 21 необходимо как можно раньше провести обследование глаз, поскольку выше перечисленные проблем можно исправить.

Пороки сердца
Около 50 процентов детей рождаются с сердечными аномалиями.
Отдельные из этих дефектов сердца являются мягкими и могут скорректироваться самостоятельно без медицинского вмешательства. Другие сердечные аномалии более тяжёлые и требуют хирургической операции или медикаментозного лечения.
Потеря слуха
Проблемы со слухом при синдроме Дауна не редкость. Отит поражает примерно от 50 до 70 процентов детей и является частой причиной утраты слуха. Глухота, присутствующая при рождении, встречается у примерно 15 процентов детей с данным генетическим расстройством.
Расстройства ЖКТ
Около 5 процентов больных младенцев будут иметь проблемы со стороны ЖКТ, такие как сужение или закупорка кишечника или заращение анального отверстия. Большинство из этих патологий можно устранить при помощи хирургии.
Отсутствие нервов в толстом кишечнике (болезнь Гиршпрунга) чаще встречается у индивидуумов с синдромом, чем у населения в целом, но все ещё довольно редко. Существует также сильная связь между целиакией и трисомией 21, что означает, что это чаще бывает у людей с этим расстройством, чем у здоровых лиц.
Эндокринные аномалии
При синдроме Дауна часто развивается гипотиреоз (недостаточное продуцирование гормонов щитовидной железы) Он может быть врождённым или приобретённым, вторичным по тиреоидиту Хашимото (аутоимунное заболевание).
Тиреодит Хашимото, который вызывает гипотиреоз, является наиболее частой болезнью щитовидной железы у пострадавших пациентов. Болезнь обычно начинает развиваться со школьного возраста. Редко, тиреоидит Хашимото приводит к гипертиреозу (избыточное продуцирование гормонов).
У лиц с синдромом есть повышенный риск развития сахарного диабета I типа.
Лейкемия
Очень редко, примерно в 1 проценте случаев, у индивидуума может развиться лейкемия. Лейкемия — это тип рака, который поражает клетки крови в костном мозге. Симптомы лейкемии включают лёгкие синяки, усталость, бледный цвет лица и необъяснимые лихорадки. Хотя лейкемия является очень серьёзным заболеванием, уровень выживаемости является высоким. Обычно она лечится химиотерапией, излучением или трансплантацией костного мозга.
Интеллектуальные проблемы

У всех лиц с синдромом есть определённая степень умственной неполноценности. Дети склонны учиться медленнее, и у них есть трудности со сложными рассуждениями и суждениями. Невозможно предсказать, какой уровень умственной отсталости будет у тех, кто родился с генетическим расстройством, хотя это станет более ясным с возрастом.
Диапазон показателя IQ- для нормального интеллекта — от 70 до 130. У человека, как считается, есть лёгкая интеллектуальная инвалидность, когда его IQ составляет от 55 до 70. У умеренно умственно отсталого человека показатель IQ от 40 до 55. У большинства пострадавших людей показатель IQ находится в интервале от лёгкой до умеренной интеллектуальной инвалидности.
Несмотря на их IQ, люди с синдромом могут учиться и развиваться на протяжении своей жизни. Этот потенциал можно максимизировать за счёт раннего вмешательства, качественного образования, поощрения, завышенных ожиданий.
Генетика
PWS связан с эпигенетическим феноменом, известным как импринтинг . Обычно плод наследует импринтированную материнскую копию генов PW и функциональную отцовскую копию генов PW. Из-за импринтинга наследуемые по материнской линии копии этих генов практически не работают, и поэтому плод полагается на экспрессию отцовских копий генов. Однако при PWS происходит мутация / делеция отцовских копий генов PW, в результате чего у плода отсутствуют функционирующие гены PW. Гены PW являются SNRPN и NDN гены, наряду с группами snoRNAs : SNORD64 , SNORD107, SNORD108 и две копии SNORD109, 29 копий SNORD116 (HBII-85) и 48 копий SNORD115 (HBII-52). Эти гены расположены на хромосоме 15, расположенной в районе 15q11-13. Эта так называемая область PWS / AS в отцовской хромосоме 15 может быть потеряна одним из нескольких генетических механизмов, что в большинстве случаев происходит в результате случайной мутации. Другие, менее распространенные механизмы включают дисомию у одного родителя , спорадические мутации , транслокации хромосом и делеции генов.
Область 15q11-13 участвует как в PWS, так и в синдроме Ангельмана (AS). В то время как PWS является результатом потери генов PW в этой области на отцовской хромосоме, потеря другого гена ( UBE3A ) в той же области на материнской хромосоме вызывает AS. PWS и AS представляют собой первые зарегистрированные случаи расстройств, связанных с импринтингом у людей.
Риск СПВ для брата или сестры пострадавшего ребенка зависит от генетического механизма, вызвавшего заболевание. Риск для братьев и сестер составляет <1%, если пораженный ребенок имеет делецию гена или однопородную дисомию, до 50%, если пораженный ребенок имеет мутацию контролирующей области импринтинга, и до 25%, если присутствует родительская хромосомная транслокация. Пренатальное тестирование возможно на любой из известных генетических механизмов.
Микроделеционный в одной семье snoRNA HBII-52 исключила его из играет важную роль в развитии этого заболевания.
Исследования модельных систем человека и мыши показали, что основной причиной СПВ является делеция 29 копий C / D box snoRNA SNORD116 (HBII-85).
Благотворительные фонды помогают приехать на лечение в Израиль
Международный отдел медицинского центра Хадасса работает в тесном контакте с ведущими благотворительными фондами, которые уже обеспечили десятки больных редкими заболеваниями детей средствами, необходимыми для прохождения лечения в МЦ Хадасса. Мы настоятельно советуем всем родителям, которые взвешивают возможность привести своих детей на лечение в Израиль, обратиться в эти организации.
- Русфонд
- World Vita
- Благотворительный фонд «Алеша»
- Благотворительный фонд «Артемка»
- Благотворительный фонд «Клуб добряков»
- Благотворительный фонд «Помогать легко»
У вас возникли вопросы?Обращайтесь к нам и получите ответ
по телефону: +972 2 560-97-99 (круглосуточно)
по электронной почте: ru-office@hadassah.org.il
или заполнив контактную форму
Диагностика ожирения
Первоначально ожирение определяется по индексу массы тела, как отношение массы тела к росту человека в метрах в квадрате. По этим расчетам можно определить степень ожирения. Затем более детально изучается состояние пациента и диагностируются причины ожирения для назначения правильного лечения. Для углубленной диагностики ожирения и выявления причин проводят:
- ЭКГ;
- УЗИ;
- МРТ;
- исследование состава тела;
- антропометрию;
- измерение артериального давления;
- исследование крови;
- анализ мочи;
- психологический анализ пациента;
- другие исследования исходя из симптомов болезни пациента.
Для составления общей картины заболевания могут потребоваться консультации эндокринолога, гинеколога, уролога, кардиолога, сомнолога, психолога, гастроэнтеролога, невролога и других специалистов.
На основе полученных данных исследований врач-диетолог определяет состояние пациента, устанавливает причины ожирения и назначает лечение, направленное на устранение причин ожирения и снижения веса.

Если говорить простым языком, что такое синдром Прадера — Вилли?
Марина Вербивская: Синдром — это ряд симптомов, и совершенно не обязательно, что все они должны быть у ребёнка. Самый часто встречающийся — гипотония: такие дети практически неподвижны первые месяцы. У моей Кристинки была сильная гипотония через несколько часов после рождения. Она лежала в отделении интенсивной терапии, а я считала, сколько раз за день она откроет глаза. А когда Кристина руку в полгода первый раз подняла, это было целое событие! Самый неприятный симптом — гиперфагия, или переедание, он появляется у ребёнка в три года или позже. У детей отсутствует чувство сытости, они постоянно хотят есть, а взрослые не понимают, в чём дело, и рады накормить.
- Дочь Марины Вербивской Кристина
- Сын Марины Амулиной Роберт
Марина Амулина: Очень опасно, когда родители не знают об этом симптоме и начинают закармливать ребёнка. У таких детей слабый метаболизм, и можно довести до ситуации, когда ожирение становится морбидным (то есть от него можно даже умереть)
Поэтому важно как можно раньше узнать о заболевании. Ещё у таких детей при рождении снижены или отсутствуют рефлексы
Когда родился Роберт, у него вообще не было ни одного жизненно важного рефлекса: он не мог ни дышать, ни сосать, ни глотать, ни откашливаться. В первый месяц жизнедеятельность поддерживалась аппаратами.
Синдром Прадера — Вилли
Это редкое наследственное генетическое нарушение, связанное с нарушением работы 15‑й хромосомы.
Характерные симптомы:
— низкая подвижность плода, позже сонливость ребёнка (гипотония);
— маленькие кисти и стопы, низкий рост;
— косоглазие;
— гиперфагия (переедание);
— искривление позвоночника, сниженная плотность костей, плохие зубы;
— сниженная функция половых желёз, позднее половое созревание, бесплодие;
— речевая задержка и задержка психического развития.
Интересные факты о синдроме
В США посчитали, сколько проживает в стране взрослых и маленьких детей с синдромом Дауна. Их оказалось 206 тысяч. Таким образом, количество в мире людей с трисомией 21-й хромосомы составляет несколько миллионов.
Информация об этой патологии доступна. В интернете есть много полезных статей, с фотографиями и пояснениями. В них простыми словами рассказывают о таком понятии, как синдром Дауна, кратко объясняют медицинскую суть проблемы и обозначают основные принципы лечения и социальной адаптации таких людей.
Благодаря этой информации общество открывает для себя даунят. В России, как и во всех цивилизованных уголках Земли, отношение к ним с каждым годом меняется в лучшую сторону. Но так было не всегда. История жестоко относилась к людям, страдающим «монгольским идиотизмом». Именно так до 1965 года называли синдром Дауна.
Природа болезни была неуточненной до 1959 года. Когда патологию стали активно изучать и проводить скрининг, многие женщины отказывались рожать при подозрении на трисомию 21-й хромосомы. Если роды происходили (беременная не обследовалась, могли врачи пропустить синдром и т. д.), от «уродливых» детей часто отказывались.
Малыши с синдромом Дауна редко воспитывались в семьях. Чаще их изолировали в специальные закрытые учреждения. В прошлом веке длительность жизни даунят была небольшой. Многие умирали в младенчестве от врожденных пороков или приобретенных тяжелых инфекций. В среднем они достигали возраста 9–10 лет.
В наши дни «солнечные дети» могут жить полноценной счастливой жизнью. Существует множество служб, организаций и обществ, которые поддерживают родителей, у которых родился дауненок. На всех стадиях роста и развития малыша семье оказывается помощь.
Учитывая мнение доктора Комаровского, можно сказать, что самым важным является начальный этап — появление ребенка на свет и принятие его своими родителями. Сейчас в интернете все чаще встречаются фото, где изображена радужная картинка — счастливая семья с малышом, имеющим синдром Дауна.
Даунята не зря стали называться «солнечные дети», так как они абсолютно беззлобные, улыбчивые и ласковые (за редким исключением). В младшем возрасте они послушны, нежны и общительны. Им требуется много внимания и заботы. Если синдром развивается в легкой форме и ребенку оказывают комплексную помощь, то в будущем он может нормально существовать в обществе.
Есть много примеров, когда человек с трисомией 21-й хромосомы добивался успеха, создавал семью, получал профессию и т. д. К известным личностям с синдромом Дауна относятся:
- Дэнни Алсабаг (австралийский актер);
- Джейми Брюэр (модель и актриса из США);
- Крис Берк (американский актер);
- Стефан Гинсз (первый, кто сыграл главную роль в фильме);
- Паскаль Дюкенн (бельгийский театральный актер);
- Андреа Фридман (актриса);
- Джудит Скотт (создатель необычных скульптур из цветных ниток);
- Пабло Пинеда (учитель и актер из Испании);
- Раймонд Ху (американский художник);
- Мэдлин Стюарт (известная модель);
- Паула Седж (актриса, общественный деятель);
- Мария Ланговая (спортсменка из Алтайского края, чемпионка мира по плаванию);
- Джек Барлоу (мальчик, выступающий с профессиональными танцорами в балетных постановках);
- Ноэлия Гарелла (аргентинская воспитательница в детском саду);
- Карен Гафни (получила докторскую степень, переплыла Ла-Манш, организовала фонд).
Ассоциации, помогающие людям с синдромом Дауна, постоянно занимаются пропагандой здорового отношения к ним, выкладывая в сети интересные факты про даунят. К сожалению, в обществе остаются и те, кто не может принять «особенных детей».
Общие сведения о синдроме Вилли-Прадера
Первое упоминание о патологии датируется 1887 годом. Лэнгдон Даун описал девочку-подростка, у которой отмечалась задержка физического развития, гипогонадизм и ожирение. Первоначально болезнь получила название «полисарция». Полноценную характеристику синдрому дали швейцарские врачи Прадер, Вилли и Лабхарт в 1956 году. Позднее в ходе глубокого изучения, доктора определили и точную локализацию генетической мутации, которая приводила к возникновению заболевания у детей. Они также связали изменения с синдромом Ангельмана. Оба расстройства провоцируются дефектом в строении 15 хромосомы. При этом в одном случае аномалия формируется в материнской копии, а в другом – в отцовской. Патология была названа синдромом Вилли-Прадера в честь врачей, внесших самый большой вклад в ее изучение. Болезнь относится к числу редких, так как ее распространенность колеблется в пределах одного случая на 10–25 тысяч новорожденных. Половой или расовой предрасположенности не установлено.

Диагностика СПВ
Диагностика болезни на начальных ее стадиях позволяет предотвратить развитие некоторых ее симптомов:
- Терапия, начатая на ранней стадии, вырабатывает у ребенка правильное пищевое поведение;
- Если до 18 месяца жизни специалисты стали корректировать соотношение гормонов роста, телосложение малыша будет развиваться правильно, как и у здорового человека.
Большие признаки, равные одному баллу:
- Периодические трудности с кормлением новорожденного;
- Задержка в познавательном развитии до 5-6 лет;
- Особые черты лица: миндалевидный разрез глаз, небольшой рот, узкая верхняя губа;
- Гипотония мышц, выявленная еще в возрасте от 1 до 3 лет;
- Изменения в строении органов репродуктивной системы;
- Развитие ожирения.
Малые признаки (0,5 балла):
- Недостаточная активность плода;
- Аномалии рефракции;
- Поражение кожных покровов;
- Пониженная пигментация радужки глаза, волос и кожного покрова;
- Густая слюна;
- Невысокий рост;
- Непропорциональные конечности;
- Проблемы со сном;
- Психические отклонения в поведении;
- Нарушение артикуляции.
Помимо вышеперечисленных критериев для точного определения диагноза следует провести кариотипирование и определить наличие различных модификаций на уровне 15 хромосомы. Также используют ДНК-маркеры и метод прометафазного анализа.
Часто патология становится заметной уже во время ультразвукового исследования беременности. Специалист замечает увеличение околоплодных вод, гипоксию плода или его нестандартное расположение. При малейших подозрениях на наличие нарушения будущей маме придется пройти перинатальную диагностику, включающую генетическое тестирование и анализ крови на уровень гонадотропина. Также для определения синдрома необходимо применить специальные молекулярно-генетические маркеры.
Дети, больные СПВ, мало двигаются, нередко крадут продукты, прячут еду и, даже несмотря на недавний перекус, постоянно остаются голодными. В таком случае появляется угроза возникновения апноэ – остановки дыхания во сне, опасной возможным смертельным исходом.
Причины
Интеллектуальная инвалидность – результат генетических заболеваний и множества других факторов, вернее, их совокупности: поведенческих, биомедицинских, социальных, образовательных.
|
Причины |
Факторы |
|||
|
биомедицинские |
социальные |
поведенческие |
образовательные |
|
|
Внутриутробное развитие плода (пренатальные) |
-возраст родителей;-заболевания у матери;-хромосомные нарушения;-врожденные синдромы |
нищенское существование матери, она подвергалась насилию, плохо питалась, не имела доступ к медуслугам |
родители употребляли алкоголь, табак, наркотики |
родители не подготовлены к появлению ребенка, когнитивно недееспособны |
|
Рождение ребенка (перинатальные) |
-недоношенность; -родовые травмы |
плохой уход за малышом |
отказ от ребенка |
отсутствие медицинского наблюдения |
|
Дальнейшая жизнь (постнатальные) |
-плохое воспитание;-травмы мозга;-дегенеративные заболевания;-эпилепсия;-менингоэнцефалит |
-бедность;-плохие взаимоотношения в семье |
-домашнее насилие, жесткость к ребенку, егоизоляция;-несоблюдение мер безопасности;-плохое поведение |
-некачественное медобслуживание и поздняя диагностика заболеваний;-недостаток воспитания;-отсутствие поддержки со стороны других членов семьи |
Конкретных «виновников», даже несмотря на довольно скрупулезные исследования и раннюю диагностику, никто точно назвать не может. Но, если проанализировать таблицу, с наибольшей вероятностью поводом появления олигофрении могут быть:
- любые генетические сбои – мутации генов, их дисфункция, хромосомные аномалии;
- наследственные отклонения развития;
- недоедание;
- инфекционные болезни матери в период беременности – сифилис, краснуха, ВИЧ, герпес, токсоплазмоз и т.п.;
- преждевременные роды;
- проблемные роды – асфиксия, механические травмы, гипоксия, асфиксия плода;
- недостаточное воспитание ребенка с рождения, родители уделяли ему мало времени;
- токсическое воздействие на плод, ведущее к поражению головного мозга – употребление родителями сильных препаратов, наркотиков, спиртных напитков, табакокурение. Сюда же относится радиация;
- инфекционные заболевания ребенка;
- травма черепа;
- болезни, затрагивающие мозг – энцефалит, коклюш, менингит, ветряная оспа;
- утопление.
Генетика
PWS связан с эпигенетическим феноменом, известным как печать. Обычно плод наследует импринтированную материнскую копию генов PW и функциональную отцовскую копию генов PW. Из-за импринтинга наследуемые по материнской линии копии этих генов практически не работают, и, следовательно, плод полагается на экспрессию отцовских копий генов. Однако в PWS происходит мутация / делеция отцовских копий генов PW, в результате чего у плода отсутствуют функционирующие гены PW. Гены PW — это SNRPN и NDN некдин гены, а также кластеры snoRNAs: SNORD64, SNORD107, SNORD108 и две копии SNORD109, 29 копий SNORD116 (HBII-85) и 48 копий SNORD115 (HBII-52). Эти гены расположены на хромосома 15 расположен в районе 15q11-13. Эта так называемая область PWS / AS в отцовской хромосоме 15 может быть потеряна одним из нескольких генетических механизмов, которые в большинстве случаев происходят в результате случайной мутации. Другие, менее распространенные механизмы включают: однопородная дисомия, спорадический мутации, хромосома транслокации, и делеции генов.
Регион 15q11-13 вовлечен как в PWS, так и в Синдром ангельмана (ТАК КАК). В то время как PWS является результатом потери генов PW в этой области отцовской хромосомы, потеря другого гена (UBE3A) в том же регионе материнской хромосомы вызывает AS. PWS и AS представляют собой первые зарегистрированные случаи расстройств, связанных с импринтингом у людей.
Риск СПВ для брата или сестры пострадавшего ребенка зависит от генетического механизма, вызвавшего заболевание. Риск для братьев и сестер составляет Пренатальное тестирование возможно при любом из известных генетических механизмов.
А микроделеция в одном семействе мяРНК HBII-52 исключил ее из основной роли в заболевании.
Исследования модельных систем человека и мыши показали удаление 29 копий C / D коробка snoRNA SNORD116 (HBII-85) является основной причиной СПВ.
Синдром Прадера-Вилли

Заболевание названо по именам исследователей, впервые описавших симптомокомплекс. Швейцарские педиатры Андреа Прадер и Генрих Вилли выделили из массы маленьких пациентов группу детей со схожими нарушениями в развитии и описали ее в середине прошлого века как отдельное заболевание.
Позднее было выявлено, что к возникновению данного синдрома приводит отсутствие или нарушение работы 7 генов (сегмент 11.2-q13) на хромосоме №15, причем из-за различия в симптомах при отсутствии или частичном нарушении выраженность заболевания может значительно варьироваться — от тяжелых случаев, диагностируемых при рождении, до стертых форм.
Изначальная причина нарушений в геноме не ясна, а вот наследуется эта патология всегда по мужской линии при наличии соответствующей генетической идеосомии у материи, и передается от отца к детям, хотя здоровые копии генов могут приводить к скрытому наследованию, и малыши с таким заболеванием рождаются и у людей без наличия синдрома.
Генетическая предрасположенность — дополнительный фактор, повышающий возраст постановки диагноза у малышей до предшкольного (и даже более позднего) обследования: внешние проявления синдрома Прадера-Вилли, достаточно нехарактерные для любой другой семьи, при явном генетическом наследовании выглядят очень логично: ребенок просто похож на папу, и внешностью, и аппетитом, и характером.
Комментарий эксперта
Екатерина Валова, психолог
Синдром Прадера-Вилли — достаточно редкая патология развития, однако опасна она не только своими проявлениями, но и неизвестностью. Характерная внешность детей, поведенческие нарушения, отставания в развитии могут сопутствовать не только множеству иных заболеваний, но и наследоваться от родителей. При такой картине понятно, что обращение к специалистам может затянуться на весь предшкольный период, а порой дети остаются без правильного диагноза и необходимого лечения, просто потому, что очень похожи на папу, а это считается нормой.
В моей практике был мальчик Володя, которого привели в развивающий центр в 8 лет с установкой «подготовить к школе»
Предшкольное тестирование весной ребенок не прошел из-за несоответствия уровня развития (память, внимание, мыслительные процессы) требованиям общеобразовательного учреждения, но родители считали, что всему виной то, что Вова не посещал детский садик, а проводил время в основном со старенькой бабушкой в деревне. Она же его и «раскормила» на козьем молоке, лишнего веса у мальчика было достаточно.
![]() Геморрой в 79% случаев убивает пациента
Геморрой в 79% случаев убивает пациента
Володя действительно «проваливал» тесты на развитие по возрасту, отличался хорошим аппетитом, отличной зрительной памятью и склонностью к играм в лего-конструкторы, иногда упрямился и «взрывался». В целом он соответствовал уровню пятилетнего ребенка. Довольно долго, месяца три после начала занятий, наши специалисты также считали причиной проблем мальчика социально-педагогическую запущенность. Но время шло, буквы не запоминались, а потом забирать Володю пришел папа с еще более выраженной внешней симптоматикой: невысокий рост, полнота, выраженная переносица, миндалевидные глаза, акромикрия и т. п. Вот с этого момента картина начала проясняться.
После длительных переговоров с мамой и уговоров папы Вова получил наконец консультацию у генетика и направление в школу специального вида. Стало ясно, что общеобразовательная школа пока не готова к обучению ребенка, отсутствует класс коррекции, и выбрасывать деньги на изучение букваря в частном центре в данный момент смысла не имеет, нужны логопеды-дефектологи, эндокринолог, специальная диета и много работы, чтобы наверстать упущенное время и предупредить сопутствующие заболевания.
Интеллект, страдающий у больных синдромом Прадера-Вилли, в случае семейного наследования заболевания осложняет коррекцию. Нередко отец не хочет принять, что и сам он болен, и ребенок нуждается в лечении. Частая смена настроения, повышенная ригидность, агрессивность, которые также являются симптомами болезни, приводят к необходимости длительной предварительной работы с членами семьи перед началом непосредственных занятий с детьми. Парам также необходима консультация генетика перед зачатием следующих детей.